



Les études du fonctionnement des enzymes, longtemps basées sur des méthodes cinétiques, ont été dominées pendant toute la fin du XXeme siècle par des techniques spectroscopiques ou structurales basées sur l'interaction matière-rayonnement: il fallait voir pour croire. Dans le même temps, le développement de méthodes numériques récompensé en 2013 par le prix Nobel de chimie allait permettre d'étudier in silico des systèmes chimiques de très grande taille. Dans un article publié dans Nature Chemistry en mars 2014, des chercheurs du laboratoire de Bioénergétique et Ingénierie des Protéines à Marseille montrent qu'il est possible de combiner les approches cinétiques et théoriques pour comprendre le mécanisme des métalloenzymes qui oxydent l'hydrogène. Il s'agit d'une collaboration avec des théoriciens italiens et anglais et des biochimistes de l'INSA à toulouse et du CEA à Saclay.
A Marseille, l'Unité de Bioénergétique et Ingénierie des Protéines (BIP) a pour champ thématique la caractérisation des chaînes respiratoires bactériennes et développe une approche pluridisciplinaire pour l'étude des relations structure-fonction dans les métalloenzymes, ainsi que leur utilisation dans le contexte des bioénergies et de l'environnement. Les hydrogénases [1], les enzymes qui catalysent la production et l'oxydation du dihydrogène (H2=2H++2e-), sont l'un des systèmes les plus étudiés au BIP.
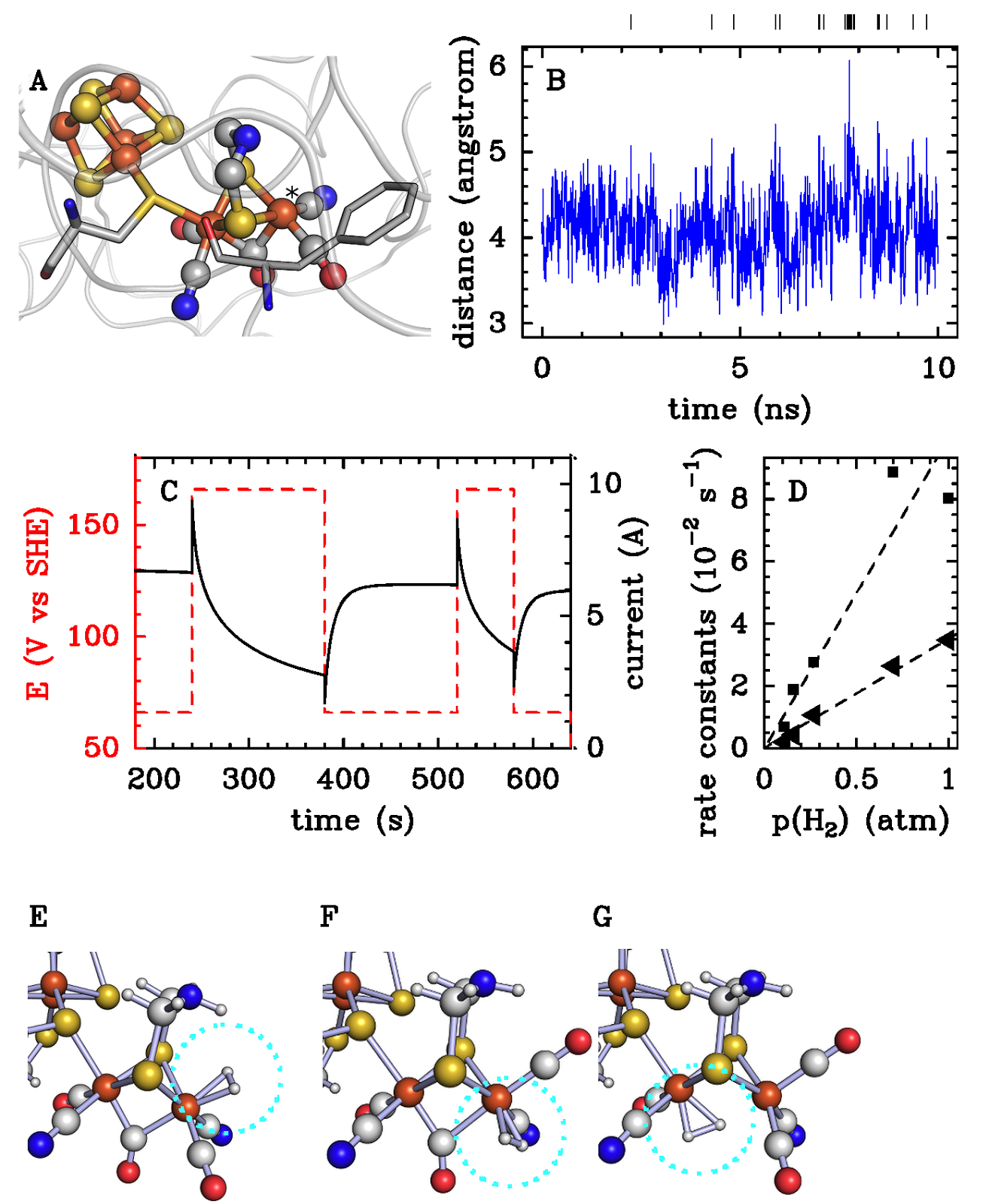
Dans les hydrogénases de type "FeFe", le dihydrogène est oxydé grâce à un site actif [Fe2CN2CO3dtma] (dtma=dithiométhylamine) représenté sur le panel A. Le dihydrogène se fixe sur le Fer "distal", marqué d'une étoile, qui est aussi la cible de l'inhibiteur O2. L'oxydation du H2 produit des protons, acceptés séquentiellement par l'azote du dtma, et des électrons qui peuvent être transférés vers une électrode, ce qui permet de mesurer la vitesse de turnover et ses variations en fonction des conditions expérimentales [2].
Lorsque le potentiel de l'électrode est changé comme indiqué sur le panel C, les variations de courant montrent que l'enzyme s'inactive à haut potentiel pour des raisons qui viennent seulement d'être éclaircies. L'hypothèse présente dans la littérature, basées sur des études de petites molécules modèles du site actif, était que l'inactivation résulte de la coordination du fer distal par l'azote du dtma; cette hypothèse est exclue par des calculs récents prenant en compte la matrice protéique autour du site actif [3].
Une information cruciale obtenue à partir des expériences d'électrochimie est que le processus d'inactivation est biphasique et que sa vitesse est proportionnelle à la concentration en H2 (panel D), ce qui démontre que l'inactivation résulte de la fixation non-productive du dihydrogène pour former deux états inactifs distincts, impliquant l’existence d’autres formes du site actif que celle qui fixe le dihydrogène dans le cycle catalytique. Les calculs de dynamique moléculaire (panel B) suggèrent que le site actif est suffisamment flexible pour autoriser le changement de position des ligands CO intrinsèques, en accord avec l'effet de la mutation de la phénlylalanine conservée (panel A) qui semble y faire obstacle. Ce mouvement rend vaquant des sites de coordination normalement occupés par les ligands CO intrinsèques, sur lesquels les calculs DFT suggèrent que le dihydrogène peut se fixer (cercles turquoises sur les panels F et G). La prédiction que le fer a une sphère de coordination complète dans les états inactifs est en accord avec l'observation expérimentale que ces derniers sont protégés contre le dioxygène [4].
C'est donc une combinaison d'informations issues de techniques très variées (mutagenèse dirigée, DFT, MD, cinétique électrochimique) qui permet ici d'élucider le mécanisme d'inactivation de l'hydrogénase et de montrer que la flexibilité du site actif autorise une chimie de coordination plus riche qu'on ne pouvait l'imaginer [4]. La comparaison de données électrochimiques et de calculs DFT nous avaient déjà permis récemment de comprendre le mécanisme par lequel l'enzyme réagit dans certaines conditions de façon irréversible avec le CO extrinsèque [5].
Références:
[1] Structure et fonction des hydrogénases, C. Léger et S. Dementin, chapitre du livre L’énergie à découvert (2013) ]
[2] V. Fourmond, C. Baffert, K. Sybirna, T. Lautier, A. Abou Hamdan, S. Dementin, P. Soucaille, I. Meynial-Salles, H. Bottin and C. Léger, “Steady-state catalytic wave-shapes for 2-electron reversible electrocatalysts and enzymes” J. Am. Chem. Soc. 125 3926 (2013). doi:10.1021/ja311607s
[3] T. Miyake, M. Bruschi, U. Cosentino, C. Baffert, V. Fourmond, C Léger, G. Moro, L. De Gioia, C. Greco. “Does the environment around the H-cluster allow coordination of the pendant amine to the catalytic iron center in [FeFe] hydrogenases? Answers from theory” J. Biol. Inorg. Chem. (2013) doi:10.1007/s00775-013-1014-4
[4] Vincent Fourmond, Claudio Greco, Kateryna Sybirna, Carole Baffert, Po-Hung Wang, Pierre Ezanno, Marco Montefiori, Maurizio Bruschi, Isabelle Meynial-Salles, Philippe Soucaille, Jochen Blumberger, Herve Bottin, Luca De Gioia and Christophe Léger, "The oxidative inactivation of FeFe hydrogenase reveals the flexibility of the H-cluster" Nature Chemistry, March 16th (2014) doi: 10.1038/nchem.1892
[5] C Baffert, L Bertini, T Lautier, C Greco, K Sybirna, P Ezanno, E Etienne, P Soucaille, P Bertrand, H Bottin, I Meynial-Salles, L De Gioia, C Léger “CO disrupts the reduced H-cluster of FeFe hydrogenase. A combined DFT and PFV study” J. Am. Chem. Soc. 133 2096-2099 (2011) doi: 10.1021/ja110627b